
Our total synthesis of
Pallambins C and D is out today in JACS. We have placed a summary of our failed
approaches and strategies in the supporting information to give the reader a glimpse
of what went into developing our synthetic route. Here we highlight a few reactions
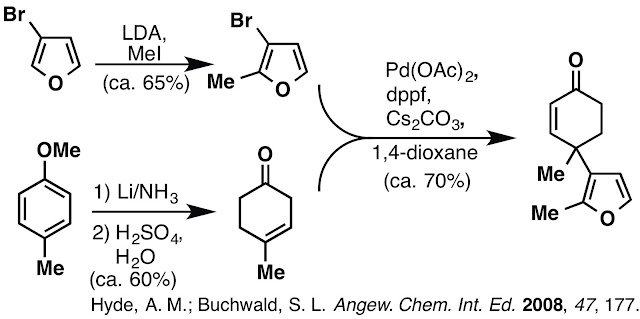
and problems encountered during this work. Our early approach began with the gamma-arylation of a beta,gamma-unsaturated ketone. We found these conditions worked
well, but as the synthesis progressed and more material was required to keep
the synthesis moving forward we noted severely decreased yields as the scale of
the reaction increased. Careful observation of the reaction mixture itself
revealed that the catalyst, ligand and base created a heterogeneous solution
which clumped up and accumulated at the liquid/gas interface of the round
bottom flask. This clumping limited the reaction to 2 mmol scale before seeing
a steep drop in yield from ca. 70% to ca. 20%. We resorted to setting up many 2
mmol scale reactions in parallel, but this was a cumbersome and not very
efficient work around the issue.
A Morton flask may have been a solution to this problem, but
we did not have one available in lab and purchasing one would not solve all the
issues with this approach. The cost of 3-bromofuran was high (to begin a
synthesis from), the stoichiometric use of Cs2CO3 was
necessary (we examined various bases) but not ideal and the enone delivered
from this method lacked an alpha-methyl group.
The sum of these issues led us to
examine an alternative Robinson annulation disconnection of the enone revealing
ethyl vinyl ketone and a furanoaldehyde. This aldehyde could arise from a
Claisen rearrangement of the corresponding allyl vinyl ether.

Although the aldehyde
was known in the literature, we found the reported 20% yield of the product to
be an unbreakable barrier for the classic Claisen rearrangement despite our best
attempts at catalysis and variation of reaction conditions. Ultimately the Eschenmoser-Claisen
rearrangement emerged as the superior reaction, but instead delivered an amide
rather than the desired aldehyde necessitating a reduction. Surprisingly, DIBAL
reduction of the amide was sluggish and only reached 50% conversation. This was
both frustrating and frightening as we did not have a way to adequately supply
material to the front line of the synthesis. Fortunately, an examination of
various reported methods led us to the use of tetramethyldisiloxane (TMDS) in
the presence of Ti(OiPr)4 to achieve the desired
reduction of the amide to an aldehyde in the same pot as the Eschemoser-Claisen
rearrangement in 75% overall yield.
Why isn’t the Robinson
annulation asymmetric?
We examined various
options in pursuit of an asymmetric synthesis with little success. Although we
were able to achieve some degree of asymmetric induction using phenylalanine,
the yield of the reaction remained low despite optimization efforts. The use of
asymmetric phase transfer conditions increased the yield of the desired enone,
but we observed a large drop in ee.

Although phase transfer
conditions did not deliver the level of ee
we desired, the increased yield was welcomed as the previous conditions for
the Robinson annulation used LiOH in isopropanol to deliver the enone in ca.
50%. We briefly revisited Buchwald’s
gamma-arylation conditions in our quest for ee. Sadly, after a small ligand screen it became apparent that this
would be a lengthy and rather expensive endeavor leading to the abandonment of
an asymmetric synthesis all together. Steps 3-11 are discussed to some degree
in either the text or supporting information and we are more than happy to
answer any question you may have regarding these steps in the comments.
Selenophenol (things I
had to work with)
As many of you know
Derek Lowe’s In The Pipeline blog sometimes features “Things I Won’t Work
With”. One particular entry
describes selenophenol (PhSeH) in all of its stinky glory. After the failure
of several endgame strategies we considered a radical cyclization approach to
D-ring formation. Attempts to install various radical precursors at the
anomeric position were surprisingly unsuccessful except for one, PhSeH. Completion
of the synthesis seemed so close at the time that we decided to go ahead and
use PhSeH. Unfortunately, installation of the selenoacetal required the use of
PhSeH as a solvent in the presence of ZnI2. We attempted to use
PhSeH in quantities up to a 1:1 solvent mixture with MeCN with no success,
leaving no option other than using PhSeH as the solvent. The selenoacetal was
installed on a model substrate (the Claisen rearrangement yield problem had not
been solved at this point) followed by installation of an alkynoate. Treatment of the alkynoate model
substrate with Bu3SnH and AIBN resulted in formation of the desired
[3.3.0]furanofuranone system on the model substrate in good yield.

To our dismay, the
success of this radical cyclization on the model substrate did not translate to
the pallambin system. We explored various methods for radical generation,
reaction concentration, rate of addition and everything else we could try to
reproduce the result of the model substrate over the course of 2 months with no
positive result. This was very tough and stinky time in lab.

Generation of PhSeH from
PhSeSePh and the subsequent distillation was a nightmare! The smell of PhSeH is
something along the lines of a dumpster filled with rotting skunks set on fire.
One afternoon while working with PhSeH a drop of the disgusting liquid fell on
one of my gloves unnoticed. I briefly removed my hand from the fume hood and
passed it near my face while reaching for something and my head was instantly
knocked back by the apocalyptic smell of PhSeH. This small but concentrated
exposure resulted in the complete loss of my sense of smell for about 10 days.
This was the absolute final straw for PhSeH and the approach was abandoned
altogether. It was quite terrifying once I passed the 7 day mark with no sense
of smell. I felt like I had made an irreversible mistake that would effect the
rest of my life. Fortunately, my sense of smell returned over the course of 3
weeks, but even a year later I’m still haunted by phantom PhSeH smells.
-Luisruben P. Martinez
Awesome description!! keep at it....♫
ReplyDeleteGoing from 10 to 11, did you see bromination at the anomeric position? Did you get exclusive retention at the brominated position?
ReplyDeleteWe did not directly observe bromination at the anomeric position upon addition of AcBr to 10. However, we did observe formation of a highly unstable enol ether possessing the desired C9 bromide if more than 1.0 equiv of AcBr was added to the reaction mixture. This enol ether was observed only through crude NMR as attempts to purify resulted in lactol formation. This is followed by a retro-aldol reaction and subsequent elimination of the C9 bromide reforming 9 likely by the sequence depicted here http://imgur.com/eL6B2r3 .
DeleteInteresting... I found it surprising that under the condition, the easily ionized and less hindered anomeric position did not get brominated first. Do you guys have any thought on that? Also, why do you think the ring closing is so stereoselective?
ReplyDelete(I don't know if the reaction you depicted can be described as retro-aldol...)
Anyway, great works! Thanks for answering my questions.
We attempted bromination of the anomeric position (C-12) as part of a separate radical cyclization endgame strategy, but never observed C-12 bromination. Instead we observed hemiacetal formation at C-12 under bromination conditions. Going from 9 to 11 I think bromination likely occurs, but the resulting bromoacetal is highly labile and can go on to further react with trace acid or water present in the reaction mixture. The yield of this reaction is 57%, with the remainder being various side products. The ring closing is selective likely due to geometric constraints allowing for only one intermediate (D) to cyclize to the desired product as depicted in figure 2. The orientation of the C-12 OMe group, although inconsequential, is guided by the anomeric effect.
DeleteGoing from 14 to15, What mechanism?
ReplyDeleteAddition of I2 to enol ether 14 leads to formation of a cyclic iodonium species on the less hindered face of the enol ether. This iodonium species is then attacked by the pre-generated tin-enolate of dimethyl malonate giving 15.
DeleteHey, Baran group! Grad student in the Romo group here wondering if you had a particular mechanistic explanation for the diastereoselectivities observed in the Mukaiyama aldol ring closure using BF3 vs TiCl4 and also the precise effect of normal addition vs reverse addition.
ReplyDeleteThe selectivity observed between BF3OEt2 and TiCl4 was discovered empirically and any answer or insight into the process is rather hand-wavy in that we have no mechanistic evidence or isolated intermediates that we can build a firm hypothesis upon. The major difference in selectivity might be attributed to the ability of TiCl4 to form a chelate with the reacting ketone and aldehyde, but again we have no evidence to draw from. Building a plastic model does not reveal an obvious lower energy conformation/orientation that would lead to a more concrete explanation.
DeleteThe observed effect of normal addition of Lewis acid vs reverse addition is reasoned as follows: Upon addition of substrate to a stoichiometric solution of Lewis acid, the desired Mukaiyama aldol reaction takes place instantly. The alkoxide resulting from attack at the C-8 ketone likely forms a stoichiometric complex with the Lewis acid preventing a nonselective retro-aldol, aldol reaction sequence at C-8. We found that an acidic work up (1M HCl, stir for 20 minutes) was required to achieve isolated yields above 25-30% for this reaction.
This comment has been removed by the author.
ReplyDeleteThis comment has been removed by the author.
ReplyDeleteAnd about Table 1, do you have any idea why only compound 18 is 1:1 diastereo mixture?
ReplyDeleteufc 205 Alvarez vs McGregor PPV Fightheld on November 12, 2016 at Madison Square Garden in New York City, New York.watch ufc 205 live stream , how to watch ufc 205 live stream online,how to be buy ufc 205 PPV.
ReplyDeleteufc 205
ufc 205 live
ufc 205 fight pass
ufc 205 live stream ppv fight
alvarez vs mcgregor
ufc 205 fight start time
ufc 205 fight card
Thanks for your kind information.So all the point which as you mention i am totally agreed .
ReplyDeletethank for sharing the link
gclub online
goldenslot
Every Boxing Fan Are Wating one the Best Boxing Fight Nigh Mayweather vs Mcgregor Showtiime PPV Boxing set one
ReplyDeleteDate: August 26, 2017,Location: T-Mobile Arena, Nevada, United States Live One Pay Pew View.Dear Are Watch One the best boxing fight live WATCH UFC LIVE STREAM PC, MAC, IPAD, IPHONE, ANDROID
Mayweather vs Mcgregor
Mayweather vs Mcgregor Live Stream
Mayweather vs Mcgregor Live Streaming
Mayweather vs Mcgregor 2017
Mcgregor vs Mayweather Live
Mcgregor vs Mayweathershow
Mcgregor vs Mayweather Fight Show
Mcgregor vs Mayweather Boxing Show
Mayweather vs Mcgregor Live Show streaming 2017