
Our recent work on
decarboxylative borylation was published in Science
today! Since Science’s new online research article format has allowed us to
include many details in the paper as well as the supplementary information, I
will avoid repeating what has been published online. Instead, I would like to
share some behind-the-scene stories.
This project originated
during a brainstorming session in Phil’s office in last May. Phil’s eyes were
beaming with excitement, describing the prospect of converting alkyl carboxylic
acids into alkyl boronates. I thought it was a workable idea but failed to
comprehend the boss man’s enthusiasm—we could turn a “C” into a “B” but I would
always want an “A”. Probably sensing my indifference, Phil told me to learn
about Velcade where the boronic acid served as a pharmacophore. I was mesmerized by the mechanism of action; I was more
thrilled by the fact that more than 15% of clinical drugs, such as chlorambucil, indomethacin and Liptor, contain the carboxylate moiety,. Could we possibly develop a method to
convert all these bioactive carboxylic acids into their bioisosteric boron
congeners? I was intrigued and embarked on the project.
{kind=link}
 |
| Alkyl Carboxylic Acids are widespread in medicine |
The initial condition screening
took several painstaking months. To make a long tough story short (see our 400+page SI),
I started the optimization from a secondary RAE, and it was found that the
preparation of MeLi and B2pin2 ate-complex, and the
addition of MgBr2 etherate were crucial. The
pre-mixed MeLi and B2pin2 provided an activated ate-complex
and allowed the coupling reaction to occur under mild conditions. The addition
of MgBr2 etherate was thought to promote the transmetallation of
boronate species onto the Ni center. However, when I applied the optimized
reaction conditions to the primary RAEs, the major products were carboxylic
acids (hydrolysis) and decarboxylative dimerization products. Interestingly, it
was found that changing the ligand from di-tBubipy
to di-MeObipy could increase the yield. We hypothesized (thanks to Pep for the
insightful discussion) that the electron-rich Ni/ligand complex could
facilitate the single electron reduction of NHPI as well as the combination of the
primary radical with the Ni center. However, further increasing the electron
density of the ligand didn’t help the reaction. At this point, I was glad that Jie
joined me in the struggle and he found that the addition of DMF increased the
yield further. Building on this finding, he came up with a set of conditions
for tertiary RAEs.


Admittedly, Miyaura
borylation (such as Fu’s elegant work) based on alkyl halides provides access
to some of our targeted boronates as well—but we’re most interested in complex
substrates such as amino acids whereby the corresponding halides are not viable
coupling partners. It was found that the decarboxylative borylation also worked
well on mono-Boc protected amino acids; however, since the electron-deficient
nature of the boron atom enabled it to form a three-membered ring with its
neighbor electron-rich nitrogen atom, such an adduct was prone to undergo C-B
bond cleavage on silica gel and I couldn’t isolate the desired product. Lisa
also found the same problem with α-oxy
carboxylic acids. In an almost illogical move, instead of going back to simpler
substrates, we started probing more challenging dipeptides. To our utter
surprise, the borylation of the dipeptide worked very well; we were able to successfully
synthesize two FDA approved drugs, velcade and ninlaro, both from simple native
peptides!
After learning about our
little success, Phil avidly asked me to do the borylation on vancomycin. After checking the calender to make sure that date was not April 1 and
spending the next half an hour drawing out the structure of our desired
borono-vancomycin, I approached Dr. Okano (Boger lab) who not only provided
me with materials, but also offered lots of insights. To our delight, we were
able to perform the borylation successfully on a vancomycin derivative.
Although this transformation did not bring about an increase in antibiotic
activity, we were excited by the reaction’s chemoselectivity.
Overall, primary (1º), secondary
(2º), tertiary (3º), benzylic (including 1º, 2º, 3º), stabilized
or non-stabilized, carboxylic acids on different ring systems, complex drugs (heteroatom-containing)
and natural products could all be
transformed to their boronic ester form smoothly using this reaction. Lisa and
Jie also found that tertiary and secondary boronate esters also could be
prepared from carboxylic acids directly when RAEs were generated in situ. The reaction conditions do
differ depending on the nature of the substrates, but we provided a Guide for Selecting Reaction Conditions in
our SI (page S31) to help the chemists who would be interested in this
chemistry. The reaction setup is not complicated (done in three stages,
including making the catalyst/ligand stock solution, preparation of the MeLi
and B2pin2 ate-complex, and mixing them together with
RAEs and MgBr2 etherate). The following video shows that Tony, our
talented high school intern and a member of our team, can independently handle
the decarboxylative borylation. To find out exactly who Tony is and see this simple reaction in action, see the video:
We were fairly content with
the reaction scope but one question eluded us: can we use this reaction to exploit the untapped medicinal potential of
boronic acids? At that moment, Phil had a conversation with “His Excellency” President Peter Schultz
about our transformation. After a few days, Dr. Arnab Chatterjee in Calibr contacted us, enquring if we could prepare for them the
following three boronic acids. The corresponding trifluoromethyl ketones are
human neutrophil elastase inhibitors that have been tested in Phase II clinic
trial for lung diseases; theoretically, the replacement of trifluoromethyl
ketone with boronic acid could enhance the activity—however,
preparations of these boronic acids are quite challenging by conventional means.
{kind=link}
 |
| Translational chemistry with the Calibr team led by Arnab Chatterjee |
Jie synthesized the first two
molecules mCBK 319 and mCBK320 using our decarboxylative borylation in a few
days, and the products were provided as single diastereomers. The initial hElastase
inhibitory evaluation (IC50) showed that both mCBK 319 and mCBK 320 were
more potent than their trifluoromethyl ketone forms, and the native peptide did
not show any activity at all. With this result, we wrapped up the study and
submitted our report to Science. It
was only later when Calibr and Jie found the third molecule mCBK 323 was the
most powerful (IC50 = 15 pM, Ki = 3.7 pM). The reviews were
encouraging; nevertheless, they also suggested that the elastase studies may be
published in a separate account as our current disclosure was too preliminary.
To me, this wasn’t a bad news at all—after all, I could end up getting two
publications from this project. However,
Phil always values quality way more than quantity. The editor, Dr Jake
Yeston, was also really accommodating and offered a second option that we
expand the study into an online research article. Phil opted for this without
any hesitation. Our colleagues from Calibr: Dr. Shan Yu, Dr. Kristen Johnson and Dr. Arnab Chatterjee worked tirelessly to complete a litany of additional
biological assays in the narrow window of several weeks.
 |
| Scalable synthesis of a future clinical candidate? |
We are also glad to report
that mCBK 323 can now be accessed on gram scales; with abundant material
supplies, in vivo biological studies
are underway.
Our decarboxylative
borylation is not without limitations. They are highlighted in the scheme
below: 1) occasionally, the purification could be a problem, both due to the excess B2pin2
and the low boiling point of some simple pinacol boronate esters; 2) thioethers
and thioesters were not well tolerated and led to low yields.
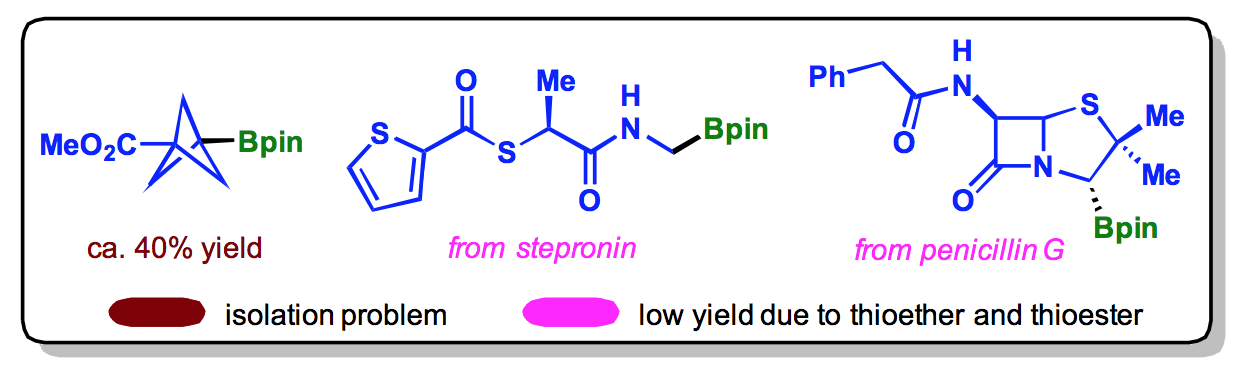 |
| Limitations |
Finally, I wanted to mention that in keeping with lab tradition, the Supporting Information is extensive. Phil told us that he wanted this document to be timeless so that if we all vanished in an earthquake or simply become too old to remember what we did, all the questions people may have and all the data needed to reproduce the reaction would be present for years to come.
As always, if there are any questions please feel free to post them here or email us!
Chao and Borylation Team.
This comment has been removed by the author.
ReplyDeleteWait! is Tony a high school student with name on science paper? That is damn impressive.
DeleteAlso, I just skimmed fast through the paper but did you try vinyl carboxylic acid? Did not see them in paper but might be pretty usefull if you can retain the cis or trans configuration (or invert it reliably)
Indeed, Tony's very impressive—we're extremely lucky to have him. The reaction with vinyl acids is a subject of ongoing investigation but for this study, it was the synthesis of alkyl boronates that interested us the most. Thank you for your question and stay tuned for updates on our future projects (e.g., maybe check back here next Wednesday?)
DeleteOh well, color me intrigued. I suppose aromatic acids are also investigated as well?
DeleteIll check for sure .)
soo Is there any update on aromatic acids you can share or should i wait for the publication? :-)
DeleteCongrats - this is really good! It beats all other alternatives how to make these peptido-boronic acids. (I remember 20 years ago, a colleague was trying to make boronic acid version of Arg, and it was an epic battle.)
ReplyDeleteMost of educational information over different kinds of blogs do not such supportive as supportive all the points of this blog. You need not to find any other platform to verify the data stated here.APC Rack AR2400
ReplyDeleteReally cool work!
ReplyDeleteYou guys should try doing this to Cetirizine - I wonder how the ether will hold up?
Keep posting stuff like this i really like it
ReplyDelete